The paper in Nature Biomedical Engineering is here: http://go.nature.com/2m7xcKu
The use of the CRISPR-Cas9 system to mediate gene editing has revolutionized the bioengineering field, as it is a very simple, highly efficient and scalable tool compared with other programmable nucleases. Hence, in a very short span of time, the CRISPR-Cas9 technology has been adopted for a broad range of applications, from basic biology to biotechnology and medicine1. Relying on the error-prone non-homologous end joining (NHEJ) DNA repair pathway, CRISPR-Cas9 has been used for efficient gene ablation. In 2014, our lab published the first paper on the use of CRISPR-Cas9 in primary human hematopoietic stem/progenitor cells for efficient ablation of a clinically relevant gene with minimal off-target mutagenesis2. Robust gene disruption in a variety of cellular systems, including human zygotes3, has been achieved successfully. However, challenges remain in the use of CRISPR-Cas9 for precision genome editing (e.g. for correction of disease-causing mutations) due to the infrequent utilization of homology-directed repair (HDR) pathway.
In recent studies, efforts have been made to improve HDR frequencies by inhibiting NHEJ4-6. Although these strategies improved HDR, such approaches may have adverse consequences given the importance of NHEJ in genome integrity maintenance7-10. We reasoned that NHEJ inhibition might not be sufficient to improve HDR to its highest levels, given that homologous recombination (HR) engagement is restricted by cell cycle phases and several factors involved in HDR could be rate limiting. Therefore, we decided to manipulate the DNA repair pathway choice through ectopic expression of several key factors involved in the HR pathway.
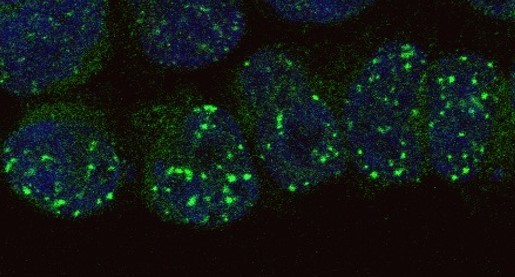
In a manuscript just published in Nature Biomedical Engineering, our group screened several DNA repair factors to find those that would be both necessary and sufficient to improve the frequency of HDR. In our paper, we report that transient co-expression of RAD52 and a dominant-negative form of 53BP1 (dn53BP1) was sufficient to increase the frequency of HDR-mediated precise gene repair at various gene loci in multiple cell types (Figure 1), including a pluripotent stem cell line derived from a patient with dyskeratosis congenita. Furthermore, we showed that this approach was applicable to multiplexing (for simultaneous modification of several genes), and could increase HDR efficacy using Cas9-nickase.

As we were modulating important DNA repair pathways for the maintenance of genomic stability, we were concerned that our approach may affect Cas9 off-target activity. To address this concern, we took advantage of an unbiased technique developed by Frederick Alt and colleagues called High-throughput Genome-wide Translocation Sequence (HTGTS)11. In collaboration with the Alt lab, our HTGTS analysis showed that Cas9 off-target activity remained unchanged as no new off-target edits were detected after transient co-expression of RAD52 and dn53BP1. This result was also confirmed independently by targeted capture deep-sequence analysis for predicted off-targets done in collaboration with Michael Talkowski’s research group.
While we were working on our paper, Lukas and colleagues12 published a study proposing a new role for 53BP1 in HDR by showing that silencing or exhausting its capacity to bind DNA induces a switch from RAD51-dependent gene conversion to RAD52-dependent single-strand annealing (SSA). This paper provided further support to our hypothesis that dn53BP1 competitively antagonizes 53BP1 to improve HDR efficiency together with RAD52, which is involved in SSA. This lead to the last figure in the manuscript providing mechanistic insight in which we corroborated our hypothesis by using a mutant dn53BP1 that does not bind to the DNA and a knockout cell line for 53BP1 that we generated in the lab. Moreover, in collaboration with the group of Tomas Kirchhausen we were also able to show that 53BP1 and dn53BP1 did not co-occupy γ-H2AX foci, indicating that dn53BP1 very effectively inhibited the recruitment of 53BP1 to the break sites.
To sum up, our data suggest a two-step model in which expression of dn53BP1 prevents the recruitment of 53BP1 to the Cas9-induced break sites, facilitating RAD52-mediated HDR using single stranded DNA donor templates (Figure 1). We hope that this new strategy will contribute to overcoming the bottleneck that still limit CRISPR-Cas9 full potential.
Written by Bruna Paulsen and Pankaj Mandal.
Our paper: Paulsen, B. S.*, Mandal, P. K.*, Frock, R. L., Boyraz, B., Yadav, R., Upadhyayula, S., Gutierrez-Martinez, P., Ebina, W., Fasth, A., Kirchhausen, T., Talkowski, M. E., Agarwal, S., Alt, F. W., Rossi, D. J. Ectopic expression of RAD52 and dn53BP1 improves homology-directed repair during CRISPR–Cas9 genome editing. Nat. Biomed. Eng. (2017) doi: 10.1038/s41551-017-0145-2.
* Equal contribution
References:
1. Hsu, P. D., Lander, E. S. & Zhang, F. Development and Applications of CRISPR-Cas9 for Genome Engineering. Cell 157, 1262–1278 (2014).
2. Mandal, P. K. et al. Efficient ablation of genes in human hematopoietic stem and effector cells using CRISPR/Cas9. Cell Stem Cell 15, 643–652 (2014).
3. Liang, P. et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell. 6, 363-372 (2015).
4. Chu, V. T. et al. Increasing the efficiency of homology-directed repair for CRISPR–Cas9-induced precise gene editing in mammalian cells. Nat. Biotechnol. 33, 543–548 (2015).
5. Maruyama, T. et al. Increasing the efficiency of precise genome editing with CRISPR–Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 33, 538–542 (2015).
6. Robert, F., Barbeau, M., Ethier, S., Dostie, J. & Pelletier, J. Pharmacological inhibition of DNA-PK stimulates Cas9-mediated genome editing. Genome Med. 7, 93 (2015).
7. Gu, Y. et al. Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity 7, 653–665 (1997).
8. Frank, K. M. et al. Late embryonic lethality and impaired V(D)J recombination in mice lacking DNA ligase IV. Nature 396, 173–177 (1998).
9. O’Driscoll, M. et al. DNA ligase IV mutations identified in patients exhibiting developmental delay and immunodeficiency. Mol. Cell 8, 1175–1185 (2001).
10. Beerman, I., Seita, J., Inlay, M. A., Weissman, I. L. & Rossi, D. J. Quiescent hematopoietic stem cells accumulate DNA damage during aging that is repaired upon entry into cell cycle. Cell Stem Cell 15, 37–50 (2014).
11. Frock, R. L. et al. Genome-wide detection of DNA double-stranded breaks induced by engineered nucleases. Nat. Biotechnol. 33, 179–186 (2014).
12. Ochs, F. et al. 53BP1 fosters fidelity of homology-directed DNA repair. Nat. Struct. Mol. Biol. 23, 714–721 (2016).



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in