"What? You take a community of bacteria and shred its DNA to study it?" - this is the typical reaction that I get when I describe metagenomics research to people outside my field. It almost sounds like fiction and I have to usually offer a few more lines of explanation of how its not that bad and that we can usually make some sense of the data.
Nevertheless, as microbiome scientists are well aware, the tools that we typically use to study microbial communities are far from perfect. The workhorse of microbiome studies, 16S rRNA sequencing, can typically resolve organisms to only the genus level - a grouping that puts together species that are separated by millions of years of evolution! Also, it reveals little in terms of what these organisms might be doing in the community and how they might influence their host or environment.
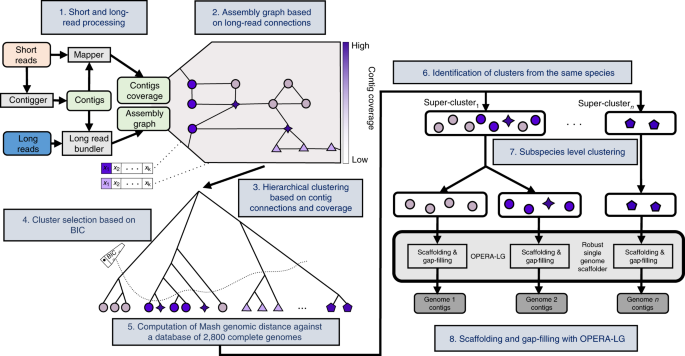
Increasingly, microbiome researchers have been using shotgun metagenomics with widely available short-read technologies for improving analysis. With the right tools, one can get species and perhaps even strain-level identification with such data. Also, the short-reads can be assembled into contigs that span a few genes, thus giving a sense of the genetic potential of the community. Assuming the genes are part of a meta-genome, researchers can then estimate the function of the community.
In reality, microbial metabolism and function happens within cells and exchange of metabolites across cells is regulated. Also, while species names and strain typing are useful proxies, the genome of a cell is the ideal identifier for its function - two genomes that differ by one gene or mutation can dramatically differ in their phenotypes (e.g. antibiotic resistance). All of this prompted us to ask a while back, "Can we study microbiomes as a collection of genomes?"
This was way back in 2012, when long read technologies were still quite expensive and we considered the use of large-fragment (5-10kbp) mate-pair libraries to improve genome reconstruction. We had just managed to generate some data on mock communities and an extremely energetic and enthusiastic intern, Senthil Muthiah, had joined the lab. Senthil relished the challenge of thinking about this new algorithmic problem. Together with Denis (the first author), we eventually formulated the core problem of using assembly connectivity (offered by the mate-pairs) and coverage to accurately cluster sequences that belonged to different genomes. Senthil used some probabilistic ideas to design an elegant and novel bayesian clustering solution. We quickly implemented a prototype and when we tested on simulated datasets, the results looked very promising. Senthil's incredibly great metagenomic assembler (aka Sigma - this is what we were calling the method then) improved assembly contiguity by an order of magnitude and introduced very few errors! This was exciting and we felt that we had something worth reporting to the world.
However, as is often the case in bioinformatics, while version 1 of Sigma worked on simulated data, it did not boost assembly as much on real datasets. What was clear to us was that real datasets had sparser connectivity information and uneven coverage and this impacted the Poisson assumption in the original implementation of Sigma. Senthil's internship ended eventually without us finding a solution to this problem. He went on to do a PhD in the University of Maryland.
The next push to the method came when another excellent intern, Mirta Dvornicic, joined the lab in 2014. By this time, long read sequencing technologies had started to become a lot more accessible and the advent of nanopore sequencing generated tremendous excitement in the field. We were hopeful that these technologies would provide the right datasets to make genome-level analysis routine for complex microbial communities. But there was still the pesky little of question of an assembler that worked! Mirta worked diligently with the conviction that a more realistic prior model (negative binomial) was what we needed to make Senthil's original idea practical. Mirta's efforts transformed our prototype version of sigma into a professional-grade version that scaled well and finally started to show promise on real datasets. Unfortunately, Mirta's internship ended in 3 months, but it showed us a exciting way forward.
The next stage of development began when the lab increasingly got involved in studies looking at the colonisation of the gut microbiome by drug-resistant bacteria. We realised that resolving genomes from metagenomes was critical for these studies to understand transmission patterns between people and environments.

Fortunately, we were also getting increasingly successful in generating long nanopore reads from stool samples. As raw error rates for this data was still greater than 5%, we knew we needed to do hybrid assemblies with Illumina data. Sigma, and the overall workflow that we built based on it (now called OPERA-MS), fit perfectly in this space.
But there were still hurdles to overcome. It turned out that metagenomic assembly in the presence of two similar strains is incredibly hard, and even with long read data, accurate clustering was tough. In came Jim Shaw, the third amazing intern who made a significant contribution to this project. Jim (along with Denis) figured out a way in which OPERA-MS could self-tune its clustering to the dataset. In addition, it could draw on information from reference genomes while still basing the assembly firmly on the read data. Jim's improvements finally made OPERA-MS produce the kind of assemblies that we believed a hybrid assembler should provide. OPERA-MS now combined high accuracy with the ability to produce near-complete genomes, even when very similar genomes were there in the dataset.
You can read all about it in the paper, but what I want to highlight is two things: a) I am glad we stuck to this project despite many hurdles and critics trying to run us down, b) Research internships can be incredible experiences and some unexpected results can come out of these efforts. Its important that we continue creating these opportunities and experiences.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in