In 2016, I crossed half of the world to join Prof. John’s Mattick’s laboratory at the Garvan Institute (Sydney, Australia) as a postdoctoral fellow. John was a strong advocate of genome-wide sequencing technologies, and was always interested in hearing about novel methods and technologies that could help us decipher the ‘big mysteries’ in biology, as he called them. At the Garvan, I met Dr. Martin Smith, who back then was postdoctoral researcher in John’s group. A few days after I joined the lab, Martin showed me a small sequencing device that seemed to be taken from a sci-fi movie: it could fit in the palm of my hand, had blue disco-like flashing lights when connected via USB, and could be directly plugged into a simple laptop. It was called ‘MinION’. I almost couldn’t believe it when I first saw this little thing in action: this pocket-sized device was able to sequence native DNA sequences without any further PCR amplification and in real-time! However, it did have some drawbacks: it was highly error-prone (30% error rate back in 2016), and it couldn’t sequence RNA molecules.

Yet, I wondered: what if we could sequence RNA with this device? I have been fascinated by RNA, and more specifically, by RNA modifications, since my early PhD years, where I studied how tRNA modifications had shaped genome structure and tRNA gene copy numbers [1]. For decades, RNA modifications had been tacitly considered a constitutive, ‘structural’ features of RNA, but this view had dramatically changed in the last years, largely due to the appearance of the first assays coupling m6A antibody immunoprecipitation to Next-Generation Sequencing (NGS), which allowed to examine for the first time, the role of m6A RNA modifications transcriptome-wide (m6A-Seq) [2,3]. Using this technique, researchers had found that m6A modifications were more widespread than previously thought, were reversible, and played pivotal roles in major biological processes, such as sex determination, cell fate transition or regulation of maternal-to-zygotic transition in vertebrate embryos [4,5,6,7]. However, m6A-Seq –-and similar technologies developed afterwards for other RNA modifications-– still presented important caveats: they could not identify how many RNA molecules were in fact modified, in which transcript isoform the modification was found in, or whether two RNA modifications were present in the same RNA molecule or were in fact mutually exclusive.
So, let’s go back again to the blue disco-like flashing device. What if we could sequence RNA with this device? And if yes, could we then use this device to detect RNA modifications directly, in native RNA molecules, and thus quantitatively? This seemed to be exactly what we needed to study the epitranscriptome! As if my prayers had been heard, we received the great news: Oxford Nanopore Technologies (ONT) would be releasing a direct RNA sequencing library preparation kit in early 2017, capable of sequencing native RNA molecules! So now the new key question was: once we would sequence the RNA molecules, how would we be able to identify RNA modifications from the raw output data produced by the nanopore sequencer?
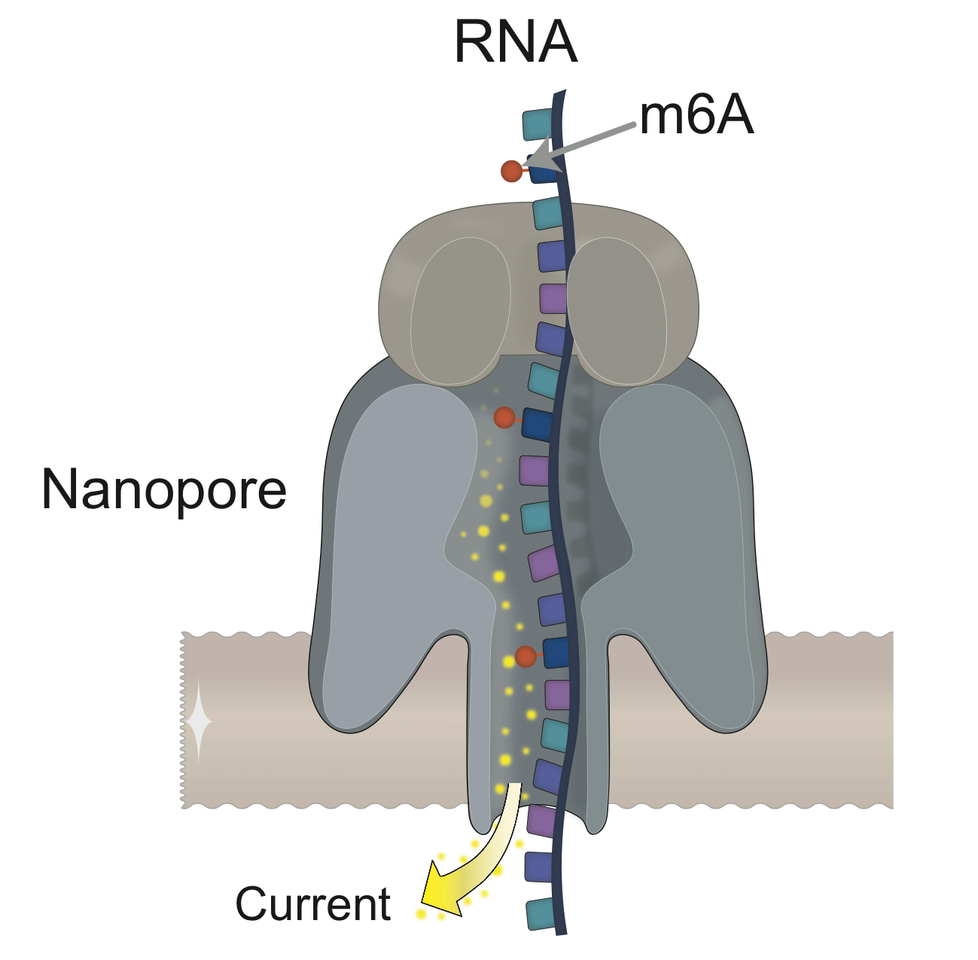
To answer this question, we must first understand how nanopore sequencing works. ONT relies on the use of membrane-embedded protein pores –-the ‘nanopores’-- which are coupled to highly sensitive ammeters that measure ionic current passing through the pore. As a molecule (DNA or RNA) moves through the pore, the sensor measures characteristic disruptions in current, which can then be used to identify the transiting nucleotides -–including modified nucleotides--. The current intensity changes are then converted into base-called nucleotide sequences using an algorithm [8].
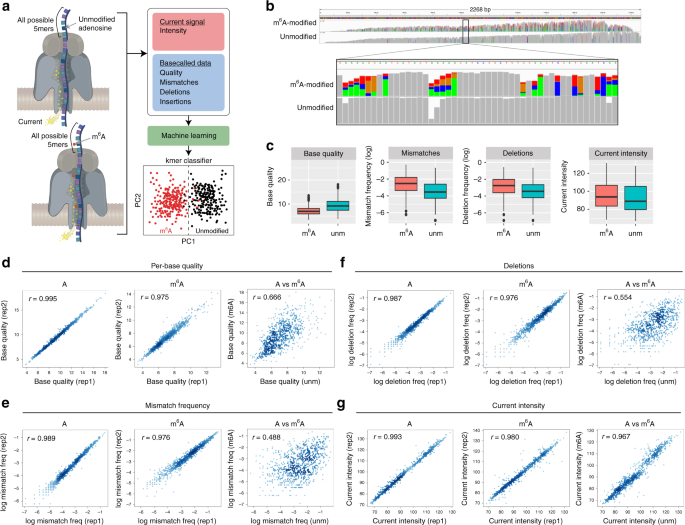
However, the algorithm that was being released by ONT would only give as output the 4 canonical letters of RNA: A, C, G and U. It thus became clear what we had to do: we had to build an algorithm to detect RNA modifications from nanopore direct RNA sequencing data, and for this, we needed proper training sets. Considering that nanopore current intensity signals are affected by its neighbouring transiting nucleotides (typically 5-mers), we designed and produced synthetic sequences that incorporated a variety of RNA modifications with all possible 5-mer sequence contexts. We termed these sequences ‘curlcakes’, due to the algorithm that was used to generate the sequences (http://cb.csail.mit.edu/cb/curlcake/). In the work that is being published today [9], we show that using m6A-modified and unmodified ‘curlcakes’, we are able to train a machine learning algorithm that can detect m6A RNA modifications with an overall accuracy of 90%. We then show that our model, trained with synthetic sequences, can be used to detect RNA modifications in mRNA sequences with an overall accuracy of 87% --using Illumina m6A-Seq datasets for orthogonal validation--.

The availability of the m6A RNA modification antibody, coupled to NGS in 2012 [2,3], completely revolutionised the epitranscriptomics field, leading to hundreds of publications that have greatly advanced our understanding of the epitranscriptome, and have put RNA modifications in the forefront of clinical studies as well as of cancer research [10,11]. I expect that the ability to detect RNA modifications in native RNA sequences, with single nucleotide and single molecule resolution, will be the key to start a second revolution in the field of epitranscriptomics, and thus reveal the secrets of this fascinating regulatory layer, capable of regulating our cells in a space-, time- and signal-dependent manner.
Acknowledgements:
Special thanks to the people and collaborators who have made this work possible, specially Huanle Liu and Oguzhan Begik who pioneered and stablished direct RNA Nanopore sequencing as well as its data analysis in our laboratory. I would also like to thank the Australian Research Council (and the anonymous reviewers) for believing in this idea even before the direct RNA sequencing kit was commercially available. I would also like to thank Martin, Chris and Schraga for a great collaboration, which has gladly been the beginning for multiple new exciting projects together.
References:
[1] Novoa, E.M., et al., A role for tRNA modifications in genome structure and codons usage. Cell, 2012. 149(1): p.202-213.
[2] Dominissini, D., et al., Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature, 2012. 485(7397): p. 201-6.
[3] Meyer, K.D., et al., Comprehensive analysis of mRNA methylation reveals enrichment in 3' UTRs and near stop codons. Cell, 2012. 149(7): p. 1635-46.
[4] Batista, P.J., et al., m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell, 2014. 15(6): p. 707-19.
[5] Lence, T., et al., m6A modulates neuronal functions and sex determination in Drosophila. Nature, 2016. 540(7632): p. 242-247.
[6] Haussmann, I.U., et al., m(6)A potentiates Sxl alternative pre-mRNA splicing for robust Drosophila sex determination. Nature, 2016. 540(7632): p. 301-304.
[7] Zhao, B.S., et al., m(6)A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature, 2017. 542(7642): p. 475-478
[8] Novoa, E.M., C.E. Mason, and J.S. Mattick, Charting the unknown epitranscriptome. Nat Rev Mol Cell Biol, 2017. 18(6): p. 339-340.
[9]. Liu, H., et al. Accurate detection of m6A RNA modifications in native RNA sequences. Nature Comm, 2019. 10: 4079.
[10] Lan, Q., et al., The critical role of RNA m6A methylation in cancer. Cancer Res 2019. 79(7): p. 1285-1292.
[11] Huang, Y., et al. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell 2019. 35(4): p. 677-691.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in