
GEM carry “effector” genes that suppress mosquito reproduction or block pathogen development in mosquitoes. Effector genes are introduced into natural mosquito populations upon GEM release and spread through wild type mosquito population by linkage to a Cas-9-based gene drive (CGD) system. Laboratory cage experiments using long-standing mosquito strains have produced encouraging CGD results. However, a flurry of recently published work has cast doubt on the viability of CGD in natural populations. requires a specific 23 base pair stretch of target DNA sequence to work. Target site variation may render CGD inoperable and individuals carrying this variation resistant to the drive. Critics argue that mosquito genomes in nature carry so much sequence variation (for An. gambiae π = 1.02%) that a significant proportion of any wild mosquito population will carry drive resistant alleles (DRAs). If a DRA is positively selected its frequency will steadily increase, thus dooming the GEM strategy to failure. Using large collections of genome sequence data from our own lab as well as the Ag1000g Consortium, we try to answer the question: How likely is it to find a CGD target site in the coding region of a mosquito gene that is conserved or only contains rare (<1%) variants which may be potential DRAs?
We examined hundreds of individual field collected mosquito genomes. Three species were included: the primary African malaria vectors, Anopheles gambiae and An. coluzzii, and the vector of the chikungunya, dengue, Yellow Fever, and Zika viruses, Aedes aegypti. We used the command line version of the tool CHOPCHOP (v2) to locate possible CRISPR/Cas9 target sites in all protein coding genes in the reference genome of each species. Sites were filtered for high specificity (no off-target sites with <4 mismatches in the genome) and GC content (30 – 70%). We produced a conservative estimate, more likely too low than too high. We then screened potential target sites for variants identified from whole-genome sequences of 1,280 field collected mosquitos. Five datasets were analyzed independently: 111 An. gambiae, 100 An. coluzzii, and 132 Ae. aegypti from our lab, along with 654 An. gambiae and 283 An. coluzzii from the Ag1000g Consortium. This protocol is summarized in Figure 1.

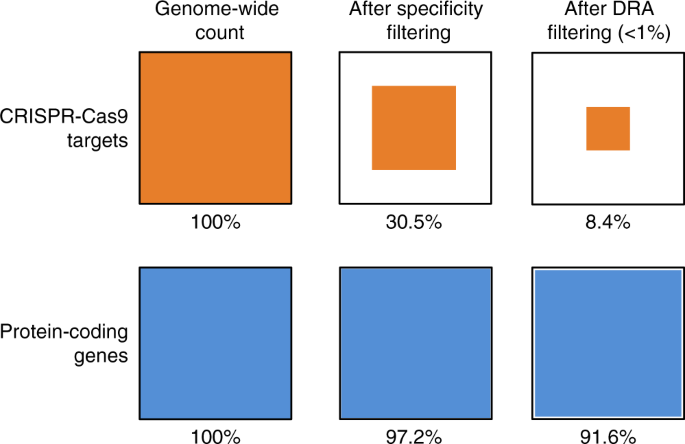
For the largest dataset (654 samples) we found that less than 3% of potential target sites had no observed variants. However, most genes contain many potential sites, so 34% of genes still contained at least one target with zero observed variants. If selection against the effector gene is weak then low frequency DRAs are likely acceptable. We found that if we set the threshold for tolerable DRA frequency at <1% then >89% of protein coding genes contain at least one useable CGD target. Target sites with no variation are rare, but those containing low frequency variants are common enough that any given gene likely contains at least one. This is good news for population modification strategies where DRAs are not under strong positive selection. In contrast, GEMs which create a large fitness cost, such as in population suppression strategies, will face significant challenges finding suitable target sites.
Vector Genetics Laboratory
Gregory Lanzaro, Ph.D,
Travis Collier, PhD
Dept. Pathology, Microbiology & Immunology
University of California
Davis, CA 95616




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in