.png "CRISPR/Cas9 methodology in human islets")
The story behind this paper started in the autumn of 2017, when I joined the Kim Lab at Stanford University School of Medicine. I was soon amazed by pancreatic islets of Langerhans: these tiny organs are responsible for maintaining whole-body glucose homeostasis, and only become functionally mature in adulthood. I was also struck by the difference between human and mouse islets -in terms of islet cellular architecture, gene expression and regulation. The Kim Lab had developed a primary organoid system -the pseudoislet platform- that enabled lentivirus-mediated shRNA ‘knockdown’ of mRNAs of interest in human primary islets. The possibility to study islet biology and genetics in primary human islets seemed unique.
I landed at the Kim Lab from a background on the -literally- opposite end of the differentiation spectrum: throughout my PhD in Buenos Aires, Argentina, I worked with mammalian embryos, and micro-manipulation of single-cell zygotes was pretty much my thing. With the advent of CRISPR/Cas9 technology, I had applied gene targeting to totipotent embryos, and the outcomes were amazing!
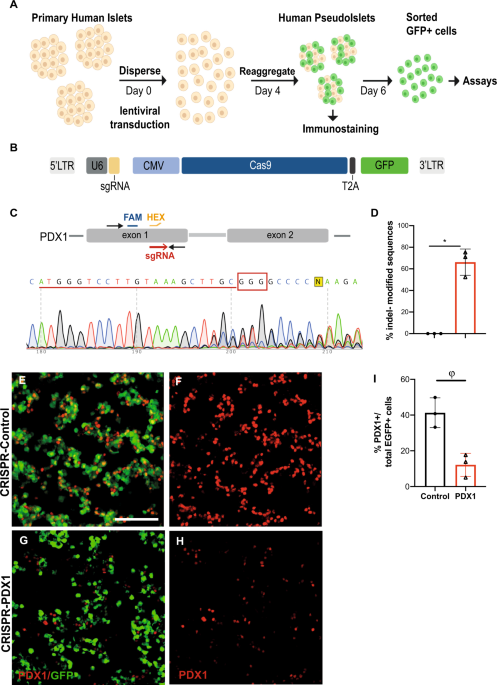
By the time I started in the Kim Lab, some reports had come out showing efficient targeting of non-dividing cells1; Seung encouraged us to apply these advances to pancreatic islet cells. There were many possibilities ahead if we could get CRISPR/Cas9 to work in primary islets, especially targeting the many non-coding presumptive regulatory regions linked by GWAS to diabetes risk. Of course, it wasn't easy in the beginning; finding the proper construct was key. After several unsuccessful attempts, I tried a lenti-construct coding for a reporter gene, Cas9 and the sgRNA, which allowed us to sort the efficiently transduced islet cells. In addition, we prolonged pseudoislets’ culture to 6 days and ultimately achieved efficient gene editing of primary human islet cells!
We demonstrated efficient targeting of exons encoding the transcription factor PDX1, with a consequent reduction of PDX1 protein and its presumptive target genes. The function of human beta cells, which synthesize and secrete insulin, was impaired both in vitro, measured in terms of excitability capacity and insulin secretion, and in vivo, after transplantation into immunocompromised mice. To test the generalizability of this approach, we designed reagents to edit KCNJ11, which encodes the KIR6.2 pore-forming subunit of KATP channels. Once again, we detected impaired islet function: in this case, the expected impairment of KATP currents.
One of the most relevant reasons to pursue genome editing in primary islets stems from the possibility of targeting the MANY SNPs (over 400 so far!) linked to increased diabetes susceptibility. Importantly, many of these SNPs fall within non-coding, presumptive regulatory regions - of which we only have information regarding theoretical targets identified through association studies, but not functional data in the proper islet context. With this in mind, we studied two presumptive regulatory regions linked to diabetes risk - in the KCNJ11/ABCC8 and SIX2/SIX3 loci - and edited them with the CRISPR system. We functionally validated the target genes of these enhancers, confirming their regulatory role, and the specificity of these, since expression of other neighboring genes was not affected. The SIX2/SIX3 locus was particularly relevant, since these transcription factors are not expressed in fetal cells (and therefore, not expressed in model cell lines), nor in mice, and close proximity of the regulatory element to the target gene impedes rigorous assessment of the interaction between the regulatory element and presumptive target genes by techniques such as pcHi-C. Ultimately, we also adapted CRISPR-on to mis-express these genes by activation of the presumptive enhancer element, further supporting our findings and interpretation.
Our results show that discrete coding and non-coding DNA elements can be targeted by CRISPR-based approaches in quiescent primary human pancreatic islet cells, and establish a paradigm for studying regulatory and genetic mechanisms linked to human diabetes risk. We are confident that our study opens the way to characterizing regulatory elements linked to diabetes risk, and that it constitutes just the beginning of multiple discoveries that should advance our understanding of diabetes susceptibility, and nominate potential therapeutic targets.
1Suzuki, K., Tsunekawa, Y., Hernandez-Benitez, R. et al. In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature 540, 144–149 (2016). https://doi.org/10.1038/nature20565



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in