Modulating the interactions between the Cas9 protein with the guide RNA-DNA complex is one way to engineer Cas9’s genome editing activity. However, the large number of residues on Cas9 in contact with the guide RNA and genomic DNA for combinatorial mutagenesis represents a huge variant space that could hardly be efficiently explored by experimental screening alone (e.g. saturated mutagenesis for 8 sites could amount to more than 25 billion Cas9 variants to be tested, let alone ‘n = >8’ number of sites). Our team has been working on formulating simplified methods for combinatorial mutagenesis in search of better Cas9 variants for therapeutic purposes. Here, we attempt to provide an applicable workflow enabling resource-efficient and high-throughput engineering of genome editors.
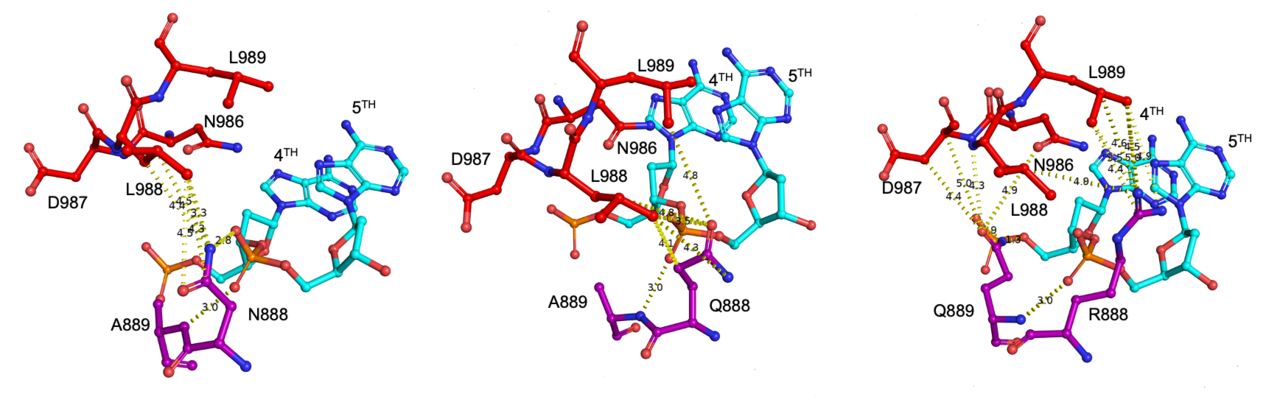
To lift the massive experimental burden, we coupled machine learning and structure-guided protein design to generate a screening library as input for virtual prediction of Cas9 activity. To do so, we applied the machine learning-assisted directed evolution (MLDE) protocol for the prediction using the previously published and our in-house mutagenesis library. We found that MLDE could identify top variants in silico and potentially reduce experimental burden by as high as 95%. The machine learning prediction was in line with the experimental screen result, showing that mutations in 2 of the 8 shortlisted sites (N888 and A889 in the WED domain) conferred enhanced activity in KKH-SaCas9 (a PAM-broaden variant of Staphylococcus aureus Cas9). We further uncovered the variant with N888R and A889Q mutations named KKH-SaCas9-Plus with enhanced gene editing activity. It showed an improvement in enzyme activity of up to 33% in endogenous loci. We also showed that this dual mutations could improve the editing efficiency of our high-fidelity SaCas9 variant, KKH-SaCas9-SAV2, and KKH-SaCas9-derived cytosine base editor. Molecular modelling shows the beneficial effect could be attributed to the increased contacts of the WED domain with L989 in the PI domain around the PAM duplex. While our work demonstrates the great potential of the machine learning-assisted approach in protein optimization, it also highlights the importance of carrying out well-designed experimental mutagenesis for generating machine learning-compatible datasets for in silico screens.
By drawing on what could be learnt by machine learning with an experimental dataset and information from the protein structure, our studies provide an approach to efficiently explore the combinatorial mutational space for the Cas9 protein optimization. Smaller Cas9 variants like Staphylococcus aureus Cas9 (SaCas9) are currently in the spotlight, as some of them have similar editing efficiency to the commonly used Streptococcus pyogenes Cas9 (SpCas9), but are smaller in size to fit in adeno-associated viral vectors to be delivered into human cells for therapies. We anticipate our approach could accelerate the engineering of more small Cas9 variants.



Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in