As the story of monoclonal antibodies and now RNA interference (RNAi) illustrates, harnessing a major discovery in biology for human health takes time. Many scientific, technical and development challenges had to be overcome and it took nearly two decades of foundational work to realize the therapeutic potential of RNAi. With four approved drugs targeting gene transcripts expressed in the liver, which are implicated in disease or disease pathophysiology, we now have a new class of innovative medicines to tackle diseases of high unmet medical need across several therapeutic areas ranging from rare to common diseases. This was only feasible thanks to several breakthroughs in the underlying technology, for which the chemistry and design of the short interfering RNAs (siRNAs) and their efficient in vivo delivery to the target cells were key. For liver delivery, this was accomplished either by encapsulating siRNAs in lipid nanoparticles1, 2 or by direct conjugation of metabolically stable siRNAs to a N-acetylgalactosamine (GalNAc)-bearing targeting ligand that is recognized by a the asialoglycoprotein receptor (ASGPR) expressed on liver hepatocytes.3 Both technologies required several rounds of optimization and refinement that greatly benefitted from the clinical experience with earlier generations of these delivery platforms.
Based on our extensive experience with conjugate-based delivery of metabolically stable and durable siRNAs to the liver, and a good understanding of the in vivo delivery requirements, we felt that we had at least some of the key tools at our disposal to broaden the scope of RNAi therapeutics to targets expressed in tissues beyond liver. We and others have been working on lipophilic conjugates for many years, intrigued by their robust activity in cell culture as well as in vivo. In fact, cholesterol conjugation has been one of the first approaches to facilitate systemic and local siRNA delivery into extrahepatic tissues including the central nervous system (CNS).4, 5 The progress with this class of conjugates has been hampered so far by a rather promiscuous delivery mechanism and concerns around safety/tolerability. Based on previous experience, we rationalized that one may take advantage of the broad cell type specificity of lipophilic conjugates, combined with a careful optimization of the lipophilicity to maximize efficiency of delivery without compromising safety.

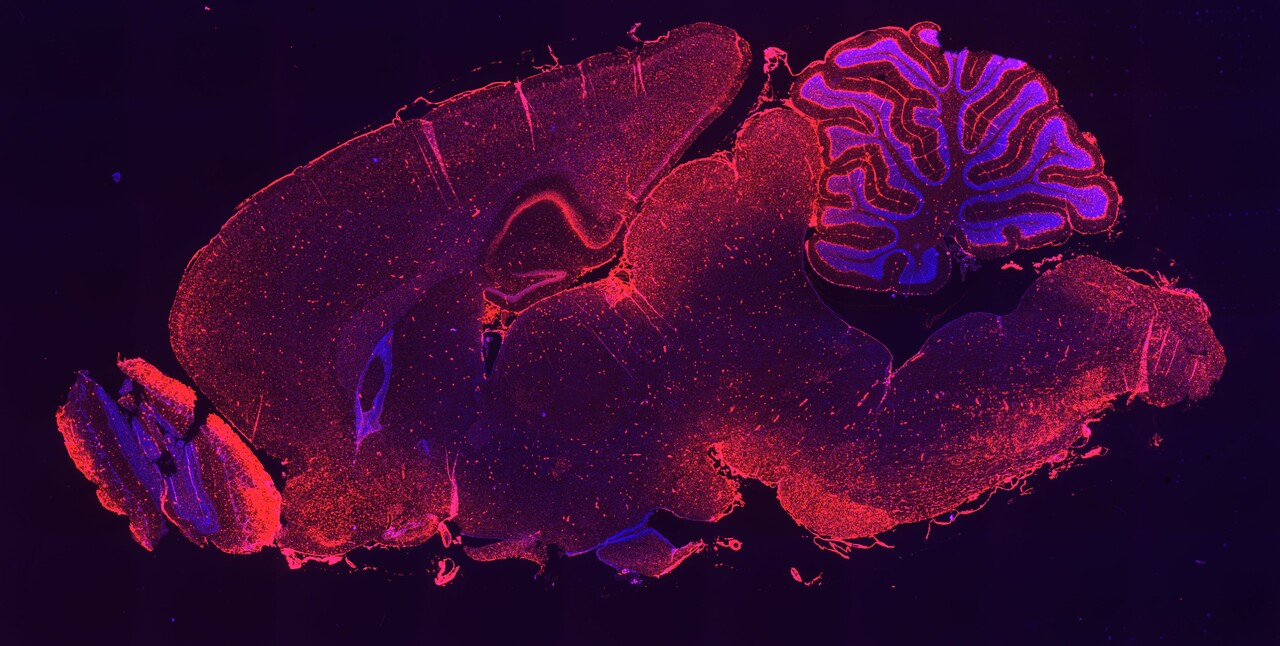
Figure 1. Example of a lipophilic-siRNA conjugate evaluated for delivery to extrahepatic tissues in this paper.
Despite several genetically validated targets and tremendous R&D efforts across industry and academia, diseases of the CNS have remained some of the most difficult to treat. However, there are few exceptions, such as nusinersen (Spinraza®), an intrathecally administered antisense oligonucleotide with a profound impact on the treatment of spinal muscular atrophy (SMA)6. Considering the high unmet medical need, we decided to try and harness the exquisite potency and durability of RNAi therapeutics for the treatment of CNS diseases. Back in 2018, after seeing the robust activity across the CNS from the first study in rat using an intrathecally-dosed lipophilic siRNA, we realized we had opened the door to a new opportunity. But we also knew it would be a long haul with many hurdles to be overcome until this technology would be ready for investigational clinical development.
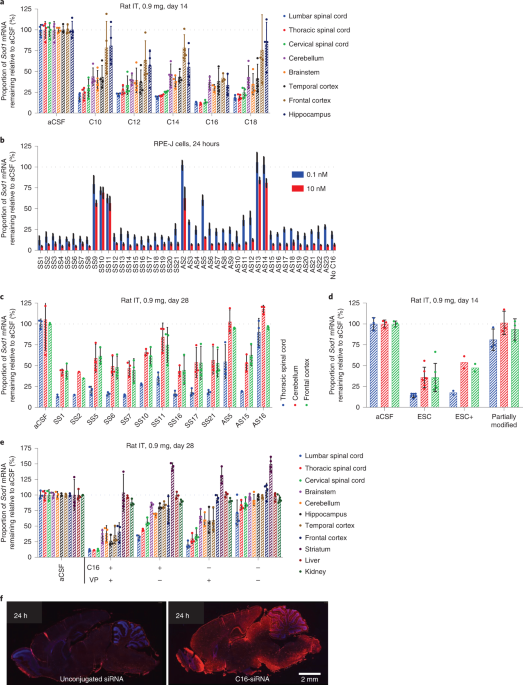
Over the following years, we embarked on a comprehensive and systematic evaluation of lipophilic siRNA conjugates in the CNS, eye and lung, which we report in this paper: https://www.nature.com/articles/s41587-022-01334-x. Starting with the CNS, we identified 2'-O-hexadecyl (C16) and other design features to arrive at a conjugate design that enables widespread siRNA distribution and durable knockdown in the spinal cord and brain following single dose into rodents (Figure 2a) and NHPs via a clinically feasible intrathecal (IT) dosing route with a favorable safety and tolerability profile.
In line with our findings in the CNS, we report broad siRNA distribution and durable activity in the eye and lung with C16-siRNA conjugates post intravitreal and intranasal administration, respectively. Figure 2b shows the broad distribution of C16-siRNA in the mouse lung at day 10 post dose. Recently, the pandemic of COVID-19 has highlighted the importance of optimizing siRNA delivery to the lung for the treatment of emergent viral respiratory diseases. While we also observed activity for the unconjugated siRNA in the eye, the C16-conjugated siRNA showed a substantial improvement in both efficacy and duration of effect. This technology could be explored for eye diseases like glaucoma and others with the potential benefit of infrequent dosing.

Figure 2. a) Superoxide dismutase 1 (SOD1) mRNA levels in different regions of brain, spinal cord and peripheral tissues after a single IT bolus injection to rats at 0.9 mg of C16-siRNA; b) siRNA distribution in the right lung of mice collected at day 10 post a single 10 mg/kg intranasal administration of C16-siRNA.
As part of the preclinical evaluation of our first development candidate for a CNS disease, ALN-APP, we were able to investigate the impact of a C16-siRNA targeting the amyloid beta precursor protein (APP) gene transcript in a mouse model of Alzheimer's disease. The treatment led to potent and durable knockdown in the CNS and ameliorated physiological and behavioral deficits. The sustained knockdown observed suggests that infrequent dosing of C16-siRNAs could be feasible in humans, which is especially important considering the IT route of administration. ALN-APP targeting APP for the treatment of early onset Alzheimer’s Disease (EOAD) and cerebral amyloid angiopathy (CAA) is now in clinical development and we are waiting to see how the preclinical results we are reporting here translate in humans for this first CNS-targeted investigational RNAi therapeutic.
- Jayaraman, M. et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chem. Int. Ed. Engl. 51, 8529-8533 (2012).
- Zimmermann, T.S. et al. RNAi-mediated gene silencing in non-human primates. Nature 441, 111-114 (2006).
- Nair, J.K. et al. Multivalent N-Acetylgalactosamine-Conjugated siRNA Localizes in Hepatocytes and Elicits Robust RNAi-Mediated Gene Silencing. J Am Chem Soc 136, 16958-16961 (2014).
- Alterman, J.F. et al. Hydrophobically Modified siRNAs Silence Huntingtin mRNA in Primary Neurons and Mouse Brain. Molecular therapy. Nucleic acids 4, e266 (2015).
- Soutschek, J. et al. Therapeutic silencing of an endogenous gene by systemic administration of modified siRNAs. Nature 432, 173-178 (2004).
- Finkel, R.S. et al. Nusinersen versus Sham Control in Infantile-Onset Spinal Muscular Atrophy. The New England journal of medicine 377, 1723-1732 (2017).




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in