
The human genome can be thought of as an extremely long word made of only 4 letters: A, C, G, T. If printed, this would fill an astounding 130 volumes. This sequence of letters is not random, rather, it has been shaped by hundreds of millions of years of evolution to encode both the recipe and the building blocks of life. A lot of these building blocks are the result of simple iterative “copy, paste and edit” processes, repurposing existing sequences and using them elsewhere in the genome for completely new functions.
Cancer is a disease of the genome originating from a single cell out of the trillions of cells in our body that started dividing too fast. During the transition to cancer, the DNA sequence of this cell can go through all the possible mishaps one can imagine: from small changes of single letters to the scrapping of whole books in the library. We refer to these modifications in our cells’ genome as somatic mutations. These mutations tell us an incredible lot about how cells become cancerous. One important and prevalent type of such mutations are called “single nucleotide variants”, which are simple substitutions of one letter by another. Even such small changes can have a dramatic impact on a cell’s behaviour. But while recognising these mutations on a genome-wide scale is crucial, it is also a complex computational task.
To date, the largest studies looking at human cancer genomes have been reading this long word by sequencing very small pieces, called “short reads”, which are limited to about 100 to 150 letters.
When sequencing cancer genomes with short reads, we try to match these reads back to the long word to know where they come from. This is the process of read alignment or mapping. But it turns out that ~10% of the reads cannot be mapped unambiguously to a single location. Rather, these reads are very similar to multiple places in the genome and can match to anything from two to tens of thousands of locations. Think of common sentences such as quotes, proverbs and epigrams, which could come from different books.
To identify somatic mutations, algorithms compare short reads taken from cancerous cells in a patient to short reads taken from their healthy cells. After read alignment, the algorithms compare these sequences at every location in the genome. The rationale is apparent: any letters that have changed in the cancer genome are potentially interesting to cancer researchers and we want to study them! Unfortunately, this is easier said than done. Many of these changes are artefacts, erroneously created by the technology. As a result, to try to compensate for errors created by any single algorithm, the current good practice is to distill a consensus from multiple independent algorithms, a process called consensus calling.
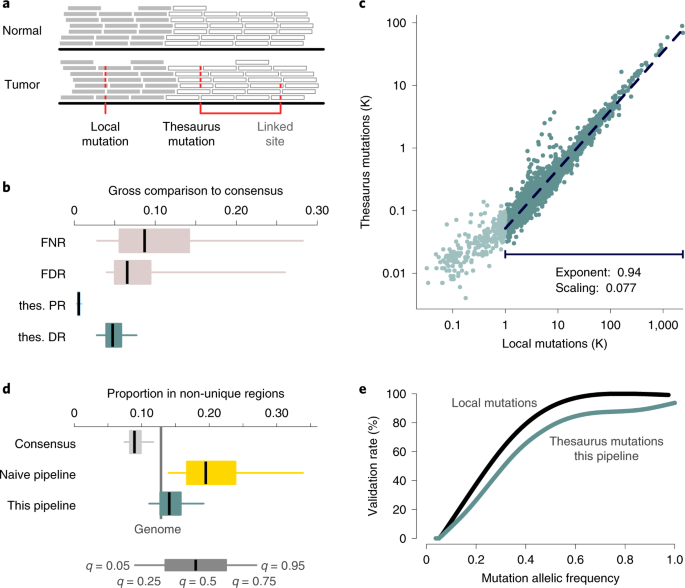
An important point is that these algorithms typically disregard reads that don't align specifically to a single location. However, this results in a blind spot for the identification of mutations from short reads. And this blindspot amounts to ~10% of the cancer genome. Imagine a book missing every tenth word!
In our study “A pan-cancer landscape of somatic mutations in non-unique regions of the human genome”, we devised a way to rescue the mutations that map to various parts of the genome. The rationale is as follows: we might not know exactly where some of the mutations should go, but we should still be able to capture their presence in the tumour and absence in the controls across their possible mapping positions. This rationale had already been exploited by Kerzendorfer, Konopka and Nijman in 2015 and forms the whole basis of this new study. I remember discussing it with Dr. Konopka on a city trip in beautiful Edinburgh in July 2016, shortly before we brought the discussion to London. He was commuting from Oxford, I from Cambridge, and we would meet at the Crick within the Van Loo lab. We got most of the ideas early in the process but it took several years to concoct the right recipe. As it happens, last year, we were finishing the work and I had come full circle and ended up back at IRIBHM, in Brussels, where Dr. Konopka and I originally met.
The innovation in our new study came from leveraging the accuracy of consensus calling in unique parts of the genome to look at the non-unique parts. We developed new computational tools that were able to keep track of information across all the possible locations short reads could map to. Crucially, unlike existing algorithms that remove the short reads which map to multiple locations, our method retains them. We tested and validated our approach on existing data relating to nerve sheath tumours, a rare type of cancer. By comparing our results against long reads collected by Dr. Demeulemeester, Dr. Verfaillie and Prof. Flanagan, we could see that our method worked effectively.
We then used the ICGC/TCGA Pan-Cancer Analysis of Whole Genomes project, a gold-mine dataset in cancer genomics, to analyse data from 2,658 tumour samples. We found millions of extra somatic mutations illuminating previous blind spots. This is a new rich resource for the scientific community, which fills apparent gaps in the mutational landscape of cancer.
We have only started to skim this new resource. A lot of genes belonging to large families with high sequence similarities have popped up in our analysis. Some that kindled our interest were members of the TRIM family, which is involved in important processes in our cells, and has been linked to cancer development. More surprisingly, we found that some sections of many known cancer genes that appeared to never mutate when assessed through classical pipelines, actually do have potential mutations.
Our pipeline did not rescue all mutations in non-unique regions. While our approach can certainly be improved, for example by combining its strengths with the power of other approaches such as reference-free k-mer approaches (akin to n-grams from books), which avoid mapping altogether, it is clear that some regions remain too difficult to look at with short-read technologies. In the near future, the adoption of long-read sequencing in combination with short reads will help us resolve many of these issues. Meanwhile, our method can be used to mine already existing short-read datasets. It could also be extended to other types of somatic mutations, such as insertions and deletions of multiple letters, as well as de-novo germline mutations, that is new mutations from one generation to the next.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in