
We come from diverse backgrounds in engineering and physics, and we were always interested in applying our skillset to solve important biological problems. We had previously collaborated on a project where we used computational tools, inspired by statistical physics, to infer a fitness landscape of HIV proteins from their global sequence data. The fitness landscape is a mapping of viral sequence data to a number which characterizes how “fit” the sequence is relative to other sequences. As a progression of that project, we were now trying to understand the viral dynamics of in-host evolution.
However, we faced a problem. The tool we used to infer the fitness landscape from global sequence data could not be applied to viral sequences collected longitudinally over time from a patient, e.g., HIV in-host evolutionary data, where the sequences are evolving due to the combination of immune pressure and viral mutations. As a first step towards addressing this problem, we started to look at evolutionary models, and how these models can be used as a starting point to infer the fitness landscape from time-series sequence data.
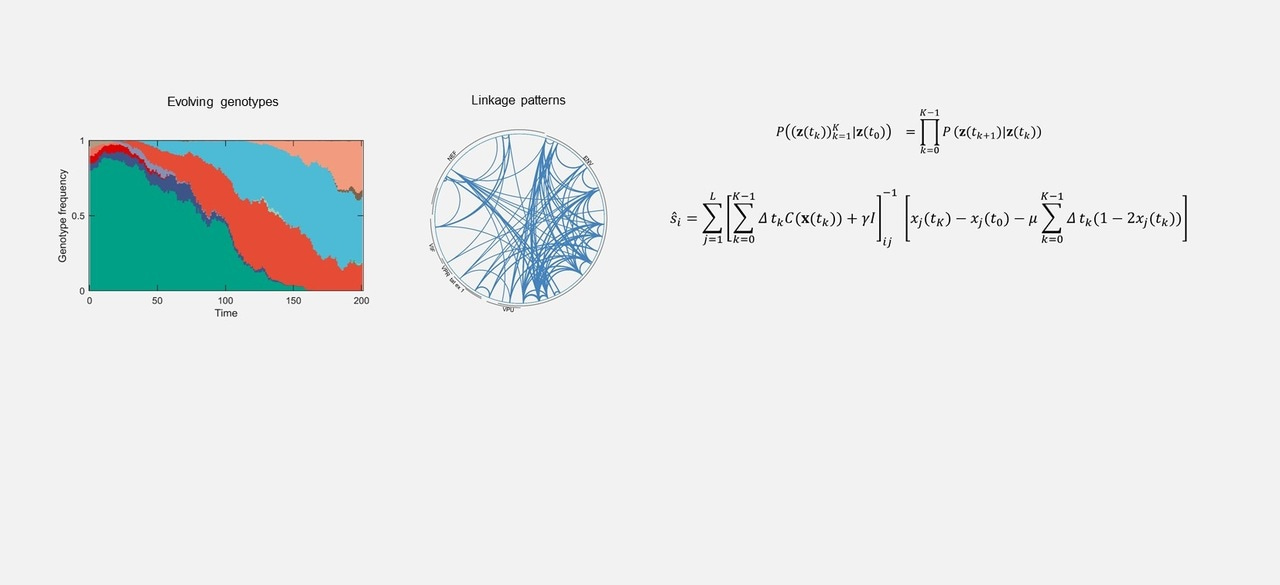
In our investigations, we realized another fundamental problem: inferring the fitness landscape based on these evolutionary models is difficult due to genetic linkage, which refers to the tendency of alleles located close in the genome to be inherited together during evolution. In other words, the effect of genetic linkage on the course of evolution is that the fate of an allele depends on its genetic background, in addition to its fitness. Linkage is a common phenomenon observed in many evolutionary settings, such as viruses and cancer. Despite this, we realized that most existing methods reported in the literature did not account for linkage effects. The handful of approaches that did account for linkage were computationally intensive and could generally only handle small systems (e.g., tens of amino acids in protein length). Inferring fitness based on evolutionary models is technically and computationally difficult, since the corresponding optimization problem is non-convex and does not scale well with the length of the protein.
Aided by our backgrounds in engineering and physics, we recognized that the fitness inference problem can be efficiently expressed as a path integral, an approach common in statistical physics. While the technical details of the approach are a bit involved, this facilitated a simple analytical solution for the estimation of the fitness effects of mutations, enabling us to analyze HIV in-host evolution data at the half-genome scale. A key feature of the method is its ability to account for linkage over large stretches of genome in a computationally efficient way, and to effectively discriminate adaptive beneficial mutations from those which are neutral or deleterious. It is also very robust to variation in the sampling parameters. Our analysis of the HIV evolutionary data of 14 individuals showed that linkage effects such as hitchhiking and clonal interference are pervasive in HIV evolution and that resolving linkage leads to substantial differences in magnitude and distribution of inferred fitness effects of mutations.
We believe our method opens new research opportunities for scientists looking to analyze evolutionary data. It may be used, for example, to study the in-host dynamics of infectious viruses other than HIV, and to potentially help distinguish driver from passenger mutations in cancer. There are also numerous opportunities for further method development by extending our proposed framework. Some extensions that our group has already considered include adaptations to apply for short-read data, and extensions of the model to account for epistasis. Other possible extensions include inference of population size, and generalizing the method for diploid models.

Figure: Mutant allele frequency trajectories exhibit complex dynamics due to linkage affects. Top left panel shows an example of observed mutant alleles trajectories, the lower three panels on the left show the trajectories of beneficial/neutral/deleterious mutants in blue/orange/red, while the right panel shows their estimated fitness values using our proposed method. Vertical lines indicate the true fitness values.




Please sign in or register for FREE
If you are a registered user on Research Communities by Springer Nature, please sign in